Agilent Laboratories Advances Bio-Analytical Measurement with Computational Biology and Informatics
|
|
March 8, 2010By Agilent Technologies Chief Technology Officer Darlene J.S. Solomon with Agilent Laboratories research and development leaders Annette Adler, Laurakay Bruhn, Robert Kincaid, Allan Kuchinsky, Nick Sampas and Anya Tsalenko Introduction Computational biology and informatics are essential to biological research at Agilent Laboratories. A good example is the development of the Agilent Oligonucleotide Array-based CGH (aCGH) DNA Microarray Platform – from initial work, primary data, visualization and integration of results to future directions.
As one of Agilent’s fastest-growing measurement solutions, aCGH microarrays identify multiple and missing pieces of chromosomes in cancer cells compared with normal cells. In a normal human genome each chromosome is present in two copies, but cancer genomes typically have multiple changes in copy numbers and rearrangements of chromosomes and pieces of chromosomes. These 1-by-3 inch glass microarray slides also can identify copy number variations (CNVs) between normal individuals1. CNVs are prevalent forms of structural variation in the genome that contribute to human genetic variability2. Computational Biology and Informatics The importance of computational biology and informatics is relatively new. Until the early 1990s, biology researchers primarily studied one or two genes at a time, often for their entire careers. High-throughput technologies like microarrays now enable biologists to study hundreds of thousands of DNA segments in a single experiment. In about 10 years the field has gone from the study of one gene at a time to all genes simultaneously. Each human cell has about 3 billion base pairs of nucleotides, including about 25,000 genes that vary in size from a few thousand to more than two million base pairs. Analyzing this almost limitless amount of data requires a computational core. Researchers use computational biology to address biological problems with disciplines such as computer science, applied mathematics and computer-simulation techniques. Specializations in this field include bio-modeling, bioinformatics, mathematical biology, computational genomics, molecular modeling, protein structure prediction, structural genomics, computational biochemistry and biophysics. Other researchers focus on informatics to build software tools that support scientists who design, conduct and collaborate on life science experiments as well as search for, store and interact with data. Informatics involves significant challenges such as visualization, human-computer design, collaborative systems, signal analysis, performance optimization, database design, and network and systems architecture. This field requires skills in the various subfields of computer science, artificial intelligence, cognitive science, information science and social science. Computational biology and informatics work together in the areas of computational creativity and expertise required to develop algorithms and computer programs. An algorithm is a detailed method for solving a problem implemented by a finite sequence of instructions that a computer will follow. A computer program is the text -- the written instructions in a specific computer language -- that will execute the instructions. It requires special artistry and deep knowledge of biology and computation for researchers (1) to outline what needs to happen in a computerized biological experiment so that each discrete step in the correct sequence will lead to the desired result, and (2) to creatively translate that sequence to the computer language so the steps are executed properly. Developing Initial Microarray Platform The team created algorithms and computer programs to guide the generation of synthetic DNA probes. At first only the genomic sequence was known. To figure out which length of probe on the microarray surface would work best, they conducted computer simulations and empirical measurements. All of the probes on the array would have to form stable double-stranded hybrids of two complementary strands of DNA under the same conditions, such as the same temperature, with good specificity and sensitivity. They calculated different metrics for each probe with computer simulations based on what could be predicted theoretically, such as thermodynamic melting temperature, binding energy, internal structure and the number of places along the entire genome that each probe was likely to bind. To test the model, the team conducted experiments using a ratio metric for sequences derived from the X chromosome. Because males have one copy and females two copies, they knew the theoretical ratio of the male to female should be primarily half on the X chromosome. They generated probes spanning the X chromosome, took measurements across male and female samples, and assigned highest scores to the probes that gave the ratio closest to the theoretical ratio. They then compared their experimental measurements with the computer simulations to see how to best predict the experimental numbers. They used these findings to construct a computer model for use in building probes for the rest of the genome. Primary Data Each Agilent microarray contains rows and rows of DNA probes that test for the abundance of a specific DNA sequence in the test sample compared to the reference sample. This abundance is measured by the color of the microarray spot when the microarray is probed by laser light. The computation team developed the processes to design the microarrays by building up the nucleotide fragments to create the probes on the slide, and to interpret the results generated by the laser scanning of the arrays. These direct results from a microarray experiment are considered primary data, and they require sophisticated software to interpret. New biological insight results from the combination of a sensitive and accurate platform and a good method for analyzing the data to understand the implications of what the experiment actually measured. Computational tools for analysis are developed in parallel with the microarray. The team builds the tools it needs for the analysis and visualization of data and transfers some of those tools to become software for customer use. Advancing the Microarray Platform for aCGH/CNV When Agilent shipped its first DNA microarrays in 2000, Labs researchers were investigating enhancements to the platform. By working with leading researchers, they had learned that higher resolution measurements of altered and missing pieces of the genome were crucial to understanding how these variations could be related to disease susceptibility and therapeutic response in different people. Agilent Labs investigated ways the company’s microarray platform could measure these natural variations. Advancing a technology platform is often a cyclic process because initial results raise questions: What if we tweaked the probes? What if we refined the algorithms or switched to a faster computing technology? Developing the aCGH/CNV platform was a cross-disciplinary project to select tens of thousands of probes of the most effective length that could span all the genes in the human genome and precisely measure the gain or loss of one small piece of a chromosome. The first aCGH array design3 had 40,000 features, and more recent designs have up to a million features on a single array. The effort required molecular biologists making the chemistry work, computational experts designing microarrays and everyone analyzing data. The team also spoke with external collaborators, thought leaders and other interested parties to understand the most important customer needs for this new application. The team was successful in developing a specialized microarray platform that uses total genomic DNA to overcome several scientific hurdles that previously impeded comparative genomic studies in cancer and other diseases. Results for aCGH include better probe design, improved aberration-calling algorithms, and ability to pick out finer and finer detail from the probes. Each slide includes up to one million features, and each feature contains millions of probes that are analyzed with computers shining laser lights hundreds of times across each feature. Visualization As with the first microarrays, no computational or visualization software tools were commercially available for aCGH/CNV to provide recipes on how to modify the platform or analyze the data in ways that would be meaningful to the worldwide scientific community. So the team developed the methods to analyze the primary data4. One method below included visualization tools for researchers to display and analyze primary data from their aCGH/CNV experiments.
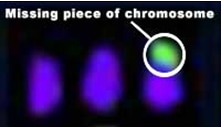
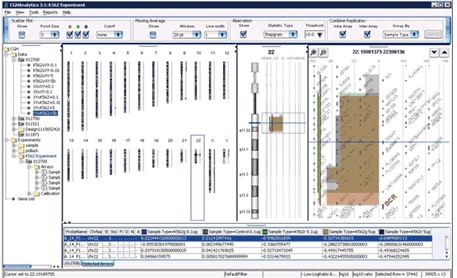
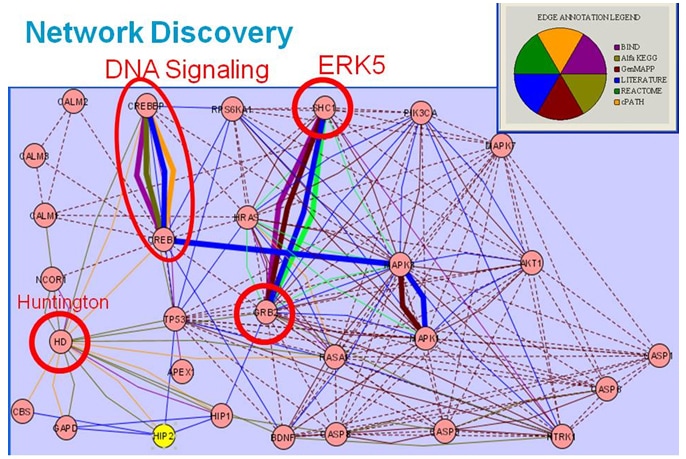
The microscopic view on the left, the previous state of the art, is part of a spectral karyotype5 image from a colon cancer cell line showing that it has three copies of chromosome 17, with a missing piece indicated in green. The view on the right developed for new aCGH measurements shows the same colon cancer cell line sample along with several other cancer cell lines missing the same piece of the chromosome. The left panel of the image shows a CGH measurement from Agilent’s aCGH platform plotted along pictures of all the human chromosomes. The middle panel shows an expanded view of chromosome 17 showing the copy number aberrations present on chromosome 17 in these cancer cell lines. The right panel shows a further expanded zoomed-in view of individual genes along chromosome 17. Integration Researchers often need to compare information and understanding obtained from high-throughput platforms such as aCHG data sets from other experiments. So biological research is becoming more collaborative as scientists seek to integrate information from multiple sources and put it into a biological context for sharing knowledge on a worldwide scale. Integration of gene expression data with networks of protein-protein interactions, for example, can reveal pathways that potentially govern disease progression. Integration with data on genetic variation might help identify people who would most likely respond to a particular therapy. A recent integration tool that encourages information sharing is Cytoscape. Cytoscape is an open source bioinformatics software platform that enables researchers to visualize molecular-interaction networks and integrate these interactions with experimental data and data from other sources, such as pathway databases and scientific literature. Putting data into a biological context facilitates understanding of the interactions between genes, proteins, and other molecules involved in biological processes. Cytoscape enables users to query biological networks to derive computational models; and to view, manipulate and analyze their data to gain biological insight. Using the Cytoscape framework, Agilent Labs collaborated with external partners using aCGH microarrays to research the integration of genetic, functional genomic and bioinformatic data for a systems biology approach to complex diseases6. They focused on a complex genetic disease, schizophrenia, to discover new candidate genes and biological pathways to aid diagnosis and treatment. The team found that a systems-biology approach offers significant promise for discovering genes of major and minor effects that collectively interact with environmental variables to produce complex disease states.
To explore relationships and illustrate genes in common between biological pathways, researchers used the Cytoscape framework, the Agilent Literature Search tool (available as a free plugin from Cytoscape– www.cytoscape.org ), protein-protein interactive databases and pathway resources. They generated a literature-based association network from approximately 450,000 PubMed abstracts retrieved for a set of disease-related queries and processed to identify approximately 39,000 bio-molecular associations among 5,400 genes. Network visualization facilitated identification of key interaction paths within the biological networks and connection points that could be therapeutic targets. The Cancer Genome Atlas Project to characterize molecular alterations in cancer is another example of integrating data from multiple sources. This collaborative effort, led by the National Cancer Institute and the National Human Genome Research Institute, has demonstrated the feasibility of using integrated genomic strategies. Scientists are developing new data, sharing it with researchers worldwide, and developing innovative bioinformatics tools and technologies to study cancer with greater precision and efficiency. Findings already are influencing treatment; investigators have reported that genetic alterations in patients with glioblastoma (a form of brain cancer) are linked with resistance to a drug that is commonly used for treatment. Using a custom-designed Agilent aCGH array, an international team of researchers at Seoul National University found that a much more accurate annotation of CNVs can be obtained for a given individual’s genome when data from high-resolution aCGH experiments is combined with whole-genome DNA sequencing data. aCGH Results Even though many cancer biologists initially were doubtful that microarrays would enable study of chromosomal variation, they now say Agilent’s aCGH array is a major technological advance that enables much higher resolution scanning of the genome and the ability to explore a whole new layer of biology. It also offers opportunities to map CNVs present in the human genome in multiple individuals across different populations. This research is a rapidly growing area because gains or losses of genomic DNA are increasingly being associated with certain genetically related diseases and developmental disorders, such as cancer, Autism and Parkinson’s. Examples of research using aCGH microarrays include the following:
Looking to the Future Researchers at Agilent Laboratories continue to work with thought leaders to integrate diverse data sets and contribute at the leading edge of advancing biological science. For example, Agilent Laboratories is collaborating with researchers at the University of California at San Francisco (UCSF). One project is combining Cytoscape and GenMAPP (Gene Map Annotator and Pathway Profiler) to enable biologists to view experimental data in a biological context that is intuitive, easy to use, and amenable to computational analysis. GeneMAPP is a computer application that UCSF designed and supports to provide a powerful modeling environment that maintains the richly annotated perspective of texbook pathway diagrams familiar to biologists. The collaboration is working to synthesize the strengths of two different perspectives on biological networks: (1) annotated pathway diagrams that molecular biologists find intuitive and (2) abstract, graphs derived from high-throughput data and amenable to large-scale network analysis.
A second project with UCSF involves development of computational tools that support community-based annotation of a protein Structure-Function linkage database. Another area of active research in Agilent Labs is visualization. Work to advance visualization tools relates to an emerging field called visual analytics. lEEE, a leading professional organization for electrical engineers and computer scientists, defines visual analytics as “the science of analytical reasoning supported by highly interactive visual interfaces. People use visual analytics tools and techniques to synthesize information into knowledge; derive insight from massive, dynamic, and often conflicting data; detect the expected and discover the unexpected; provide timely, defensible, and understandable assessments; and communicate assessments effectively for action.” This interdisciplinary science includes statistics, mathematics, knowledge representation, management and discovery technologies, cognitive and perceptual sciences, decision sciences and more. Although all sciences are improving the ability to collect and analyze information with computation and informatics, new tools are required to analyze massive, complex, incomplete and uncertain worldwide information. IEEE recognizes this challenge and in 2006 founded the Symposium on Visual Analytics, Science and Technology. It focuses on the R&D agenda for visual analytics developed under the leadership of the Pacific Northwest National Laboratory to define the directions and priorities for future R&D programs focused on visual analytics tools. Finally, the computational biologists at Agilent Labs are working on how to do ‘joint analysis’ or true integration of large sets of data of different types to provide insight into cellular mechanisms and processes. An example of this has been the joint analysis of aCGH data with transcriptional profiling data to understand how specific chromosomal aberrations affect gene expression. Agilent Labs has done work in this arena in the past, and Agilent seeks to offer help to customers who need solutions to their problems of enormous amounts of two or more data types with little idea of how to combine these into biological or clinical understanding. Researchers at Agilent Laboratories are continuing to advance computational biology and informatics through multidisciplinary research to answer ever-new and increasing complex biological questions.
|
|||||||||||||||||||||||||||||||